|
|

|
|
|
Accuracy Of Binding Energy Estimations
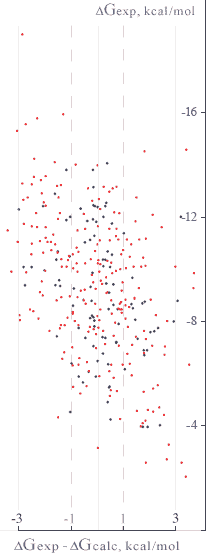
Lead Finder uses a special type of scoring function to calculate free energy ofprotein-ligand binding. This scoring function includes all energy terms (described in Scoring function section) scaled with a set of coefficients that have been adjusted to fit experimentally measured binding energies.
A setof 330 protein-ligand complexes with experimentally measured binding constants
and available3D structures was used for the current
benchmarking study, out of which 100 complexes (the training set) were used to calibrate the scoring
function. The RMSD between the calculated and experimentally determined binding energies was found to be
1.24 kcal/mol for the training set and
1.5 kcal/mol for the entire set
of 330 protein-ligand complexes.
About50% of all protein-ligand
complexes fell into a±1 kcal/mol bin
and79% fell into a±2 kcal/mol bin
ofdG prediction accuracy.
The data illustrate that Lead Finder has exceeded other docking
software
1,
2,
3
in the accuracy of binding energy estimation.
Set Of Protein-Ligand Complexes For Benchmarking Studies
To make current parameterization and benchmarking studies more meaningful,
protein-ligand
complexes with known3D structure
and experimentally measured binding constants were chosen on the basis of maximum diversity
of ligand's physicochemical properties (molecular weight (Mw),
clogP, number of hydrogen bond donors (dHB) and acceptors (aHB), net charge) and range
of protein - ligand binding energies. Physicochemical properties of selected ligands
substantially cover chemical subspace satisfying
Lipinsky's Rule of Five
4.
However, ligands spanning beyond this subspace (57 compounds with Mw>500; 31 compounds
with clogP>5; 89 compounds with (dHB+aHB)>10;
41 compounds with dHB>5) were also generously represented,
which is very important according to recent findings that physicochemical properties of drugs
are not tightly restrictedby Lipinsky rule
5.
These facts support broad applicability
of Lead Finder
for binding energy calculations.
Speed Of Binding Energy Calculations
Lead Finder provides
favorable speed of binding energy calculations compared to such methods as
free energy perturbation
or linear interaction energy that requires long molecular dynamical
simulations and individual treatment of each particular task.
For the set of studied
330 protein-ligand
complexes, the average time for binding calculations
withLead Finder
was less than one second per compound on a standard single-processor Pentium PC.
|
|
|
E-mail: info@biomoltech.com |
Phone: +1(416)238-1263 |
Fax: +1(416)352-6117 Mailing address: 226 York Mills Rd, Toronto, Ontario M2L 1L1, Canada |